

Abstract
Human migration has reached an unprecedented level. Progressive globalization has integrated people and facilitated the introduction of deleterious genes into populations in which they were originally absent, thus impacting negatively on public health. A great challenge encountered by clinicians in the era of "migration hematology" lies in the hemoglobinopathies, a group of conditions representing the commonest, life-threatening, monogenic disorders globally. Of the ±400 000 newborn children affected annually, ~65% have sickle cell disease (SCD), a disorder that has spread far beyond its origins because of slave trade and contemporary population movements. More than 70% of all global SCD cases are due to autosomal recessive homozygous inheritance of a βS-mutation (missense Glu6Val) in the HBB gene, creating sickle hemoglobin (HbS), a structural variant of adult hemoglobin (HbA) and causing the most severe form of SCD, sickle cell anemia (SCA). Other forms result from inheritance of HbS in combination with different mutations, most commonly a second structural β-globin variant, βC (S/C) or one of many that lead to decreased β-globin synthesis (S/β-thalassemia). Clinical consequences are due to polymerization of deoxygenated HbS and its deleterious effects on erythrocytes. It is a debilitating syndrome characterized by chronic hemolysis and vaso-occlusive crises, manifesting as acute painful episodes, organ infarction and eventual multi-organ damage.
Early diagnosis of SCD is vital so that appropriate management can be initiated timeously. Current diagnostic tests include hemoglobin electrophoresis (HE), high performance liquid chromatography (HPLC), isoelectric focusing (IEF) and DNA analysis. These specialized expensive modalities have been successfully incorporated into antenatal/neonatal screening programmes of some high-income countries such as in the UK. However, implementation of similar initiatives in various other regions has been difficult due to practical, political and economic challenges.
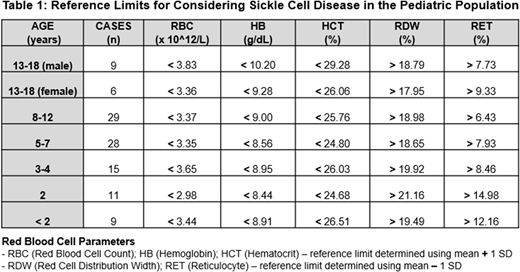
The present study outlines a simple algorithm for use in the pediatric population to identify children at risk for SCD. Residual EDTA-anticoagulated capillary blood samples were analyzed in a tertiary hospital laboratory using an automated benchtop hematology instrument (Sysmex XN-450) that provides a CBC, differential white cell count and reticulocyte percentage (RET%). Patients with known SCD (85% SCA), confirmed by HE and HPLC, presenting for routine out-patient clinic review, were included (n=107; 1 month to 18 years old; 54% male). Samples were processed within 12 hours of collection and those who had received blood transfusions in the preceding 12 weeks were excluded.
The mean ± SD was determined for each CBC parameter and RET%. Of these, 5 parameters were used to establish an algorithm with reference limits specific for each age category of patients with SCD (Table 1). When compared to the population-based normal reference ranges, these limits were statistically significantly different (p<0.05). The algorithm was validated against 107 random blood results previously obtained on routine follow up of pediatric patients with SCD. If a single outlying parameter was allowed, the algorithm demonstrated a sensitivity of 95.3% for SCD. The 4.7% of outlying cases (n=5) exhibited severe microcytosis and had a coexisting thalassemia component. The sensitivity with a complete match, i.e. 5/5 parameters, was 86.9%. Other CBC parameters were not informative.
To combat the burden of SCD early and accurate diagnosis is critical. The algorithm described here is a cost-effective and sensitive filtering tool that can quickly be used to identify children at risk for SCD at any age and exclude those who do not require definitive testing, thereby limiting unnecessary expenditure. It will be beneficial in resource-constrained low-income countries; regions where SCD is not historically present and clinical suspicion is low; or in scenarios where it is difficult to obtain a medical history due to a language barrier and/or health-related stigma. Since using the algorithm only requires a CBC and RET%, it is suitable for application at any healthcare level. Assessment may be conveniently conducted in line with a child's immunization schedule prior to symptomatic presentation, and if combined with the Mentzer index and a family origin questionnaire, the sensitivity can be enhanced.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal